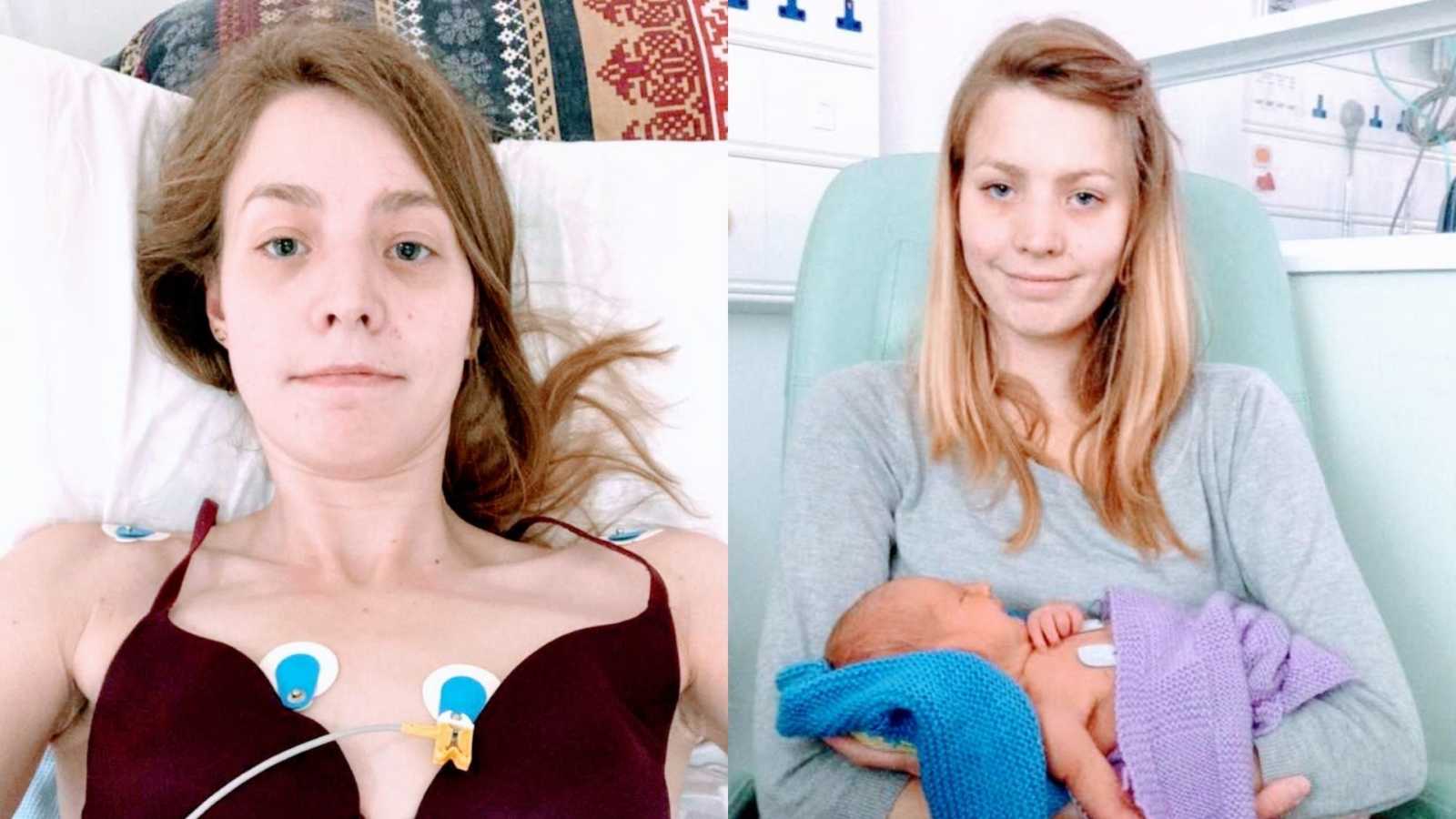
“My name is Abi Halstead. I am a married mother of three small children, living in rural England and helping run a small business with my husband. To me, my life is normal (maybe a little busy), although to an outsider it must seem anything but. On a daily basis, I inhale four nebulizers, perform two physiotherapy sessions, consume two high-calorie prescribed supplement drinks, and swallow up to 51 pills. I do all of this in order to make my faulty organs function.

When I was 4 years old, I was diagnosed with cystic fibrosis, a rare life-limiting multi-system genetic disease. Normally CF is diagnosed within the first few weeks of life, but unfortunately for me and my parents, I slipped through the net. This was not because I was a well child – far from it. I was extremely malnourished, very small, and suffered from two rectal prolapses in my first 2 years of life; a direct result of constant awful diarrhea. Despite this, I was always hungry. My parents struggled to keep me out of kitchen cupboards as I tried to find a way to fill myself. I cried a lot and was generally very angry. My parents knew something was wrong, but our GP at the time was unwilling to help.
By the time I had reached school age, my feet hadn’t grown for over a year and my stomach was distended. Fortunately, my parents were able to take me to the school doctor, who agreed with them something was seriously wrong and sent me to the hospital for tests. I was tested for diabetes and cystic fibrosis during the appointment, and my parents took me home to anxiously await the results. A few days later, my parents received a phone call from the man who would become my CF consultant, informing them their middle child and only daughter had been born with cystic fibrosis. My parents took me out of school for the day and straight to the hospital. Here we were introduced to a whole team of medical staff – a doctor, nurses, a physio, and a dietician. I underwent extensive scans and testing, while my parents had CF explained to them in detail, along with a rundown of all the new medication I would need to start taking.
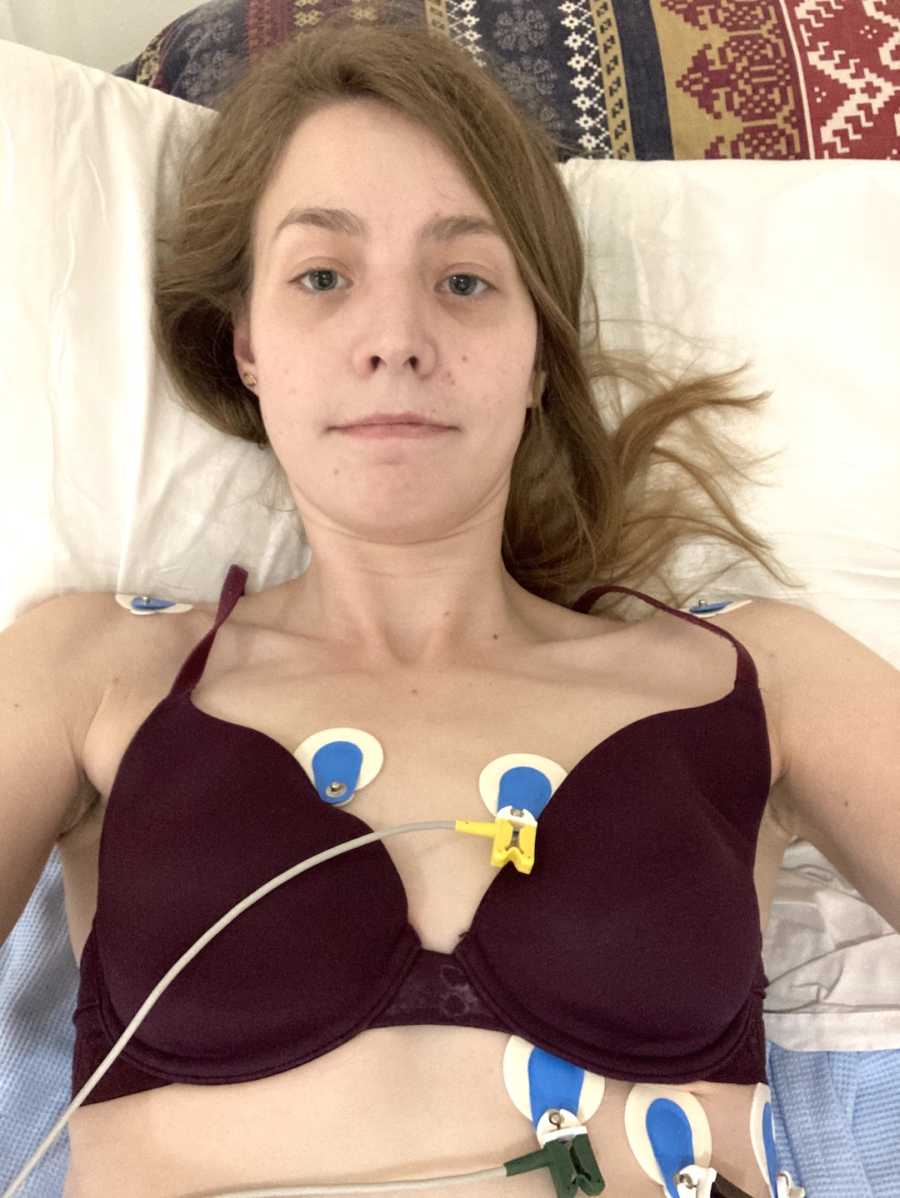
Naturally, this was a devastating blow for them. Back in 1995, the life expectancy of a CF patient was only 21 years, and my delayed diagnosis seemed likely to have lowered that. My parents were suddenly faced with raising a child who wouldn’t live long into adulthood, possibly not even seeing teenage years. However, despite their shock and upset, my parents decided from the very beginning life must go on as normal. They began my strict regiment of pills and two 20-minute physio sessions a day but continued to treat me the same as my brothers in every other way. Although the initial prognosis had been grim, my childhood and teenage years were incredibly healthy, happy, and very ‘normal.’ My medication, physio, and supplements kept me well and allowed me to grow. I worked hard at school, went to university, practiced judo and swimming, and spent summers climbing mountains and camping.
At the age of 15, I met my now husband, John. We dated for several years before tying the knot just before my 20th birthday when he was 23. Even at this point in my life my CF wasn’t really affecting me. When I was 22, however, things began to change.

Cystic fibrosis is a genetic disease caused by a tiny fault in a protein made in every epithelial (skin) cell in the body. The purpose of the protein is to pass salt and water correctly, which is what makes the mucus in a non-CF person’s body thin, slippy and effective. In a CF patient, the balance of salt and water is wrong, so the mucus becomes thick, sticky and begins to clog up organs. Epithelial cells are found in the lungs, pancreas, liver, gall bladder, intestines, sinuses and reproductive organs. All of these begin to block up in a CF patient, causing a wide range of problems. In the digestive system, most patients (myself included) have blocked pancreatic ducts and cannot digest fat properly. The mucus also slows down the speed of the intestines, and also hugely decreases their surface area, making it hard to get enough nutrients into the body. In the lungs the mucus is normally used for trapping bacteria and viruses before transporting them (cocooned in slippy clear mucus) out and down to the stomach for destruction. In CF, the mucus is a different chemical balance and consistency. This makes CF lungs the perfect environment for bacteria to get in, settle down and start throwing wild parties – wrecking the joint and breeding all over the place. This results in CF patients becoming hosts to long term, deeply unpleasant bacteria. These infections are constant although worsen regularly, which requires patients to go to hospital frequently for intense courses of IV antibiotics.
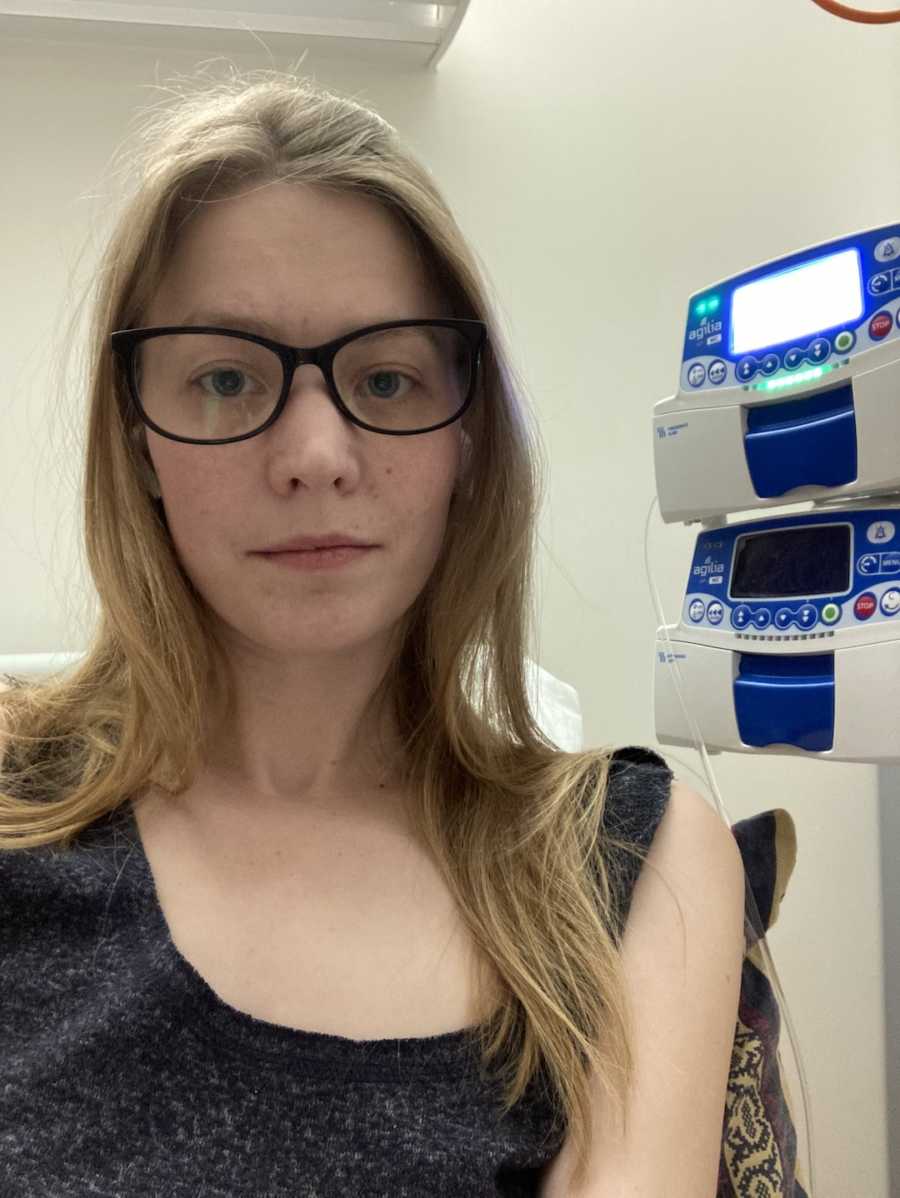
When I was 16, I caught my first nasty CF bug. It’s called burkholderia gladioli and has been with me ever since. At first I was horrified to have caught it and very afraid I would soon die from it. However, it appeared at first to be lying dormant in my lungs, not damaging anything. After a few months of stress and panic I acclimated to it being in my body and stopped worrying about it. Sadly, as time has gone on, it has woken up and become more and more aggressive. When I was 22, I became unwell enough with my CF to need a hospital stay for IV treatment. This was very unusual — most CF patients have had a lot of IV experience from very early on in their lives. Reaching 22 without intervention had been quite a feat.

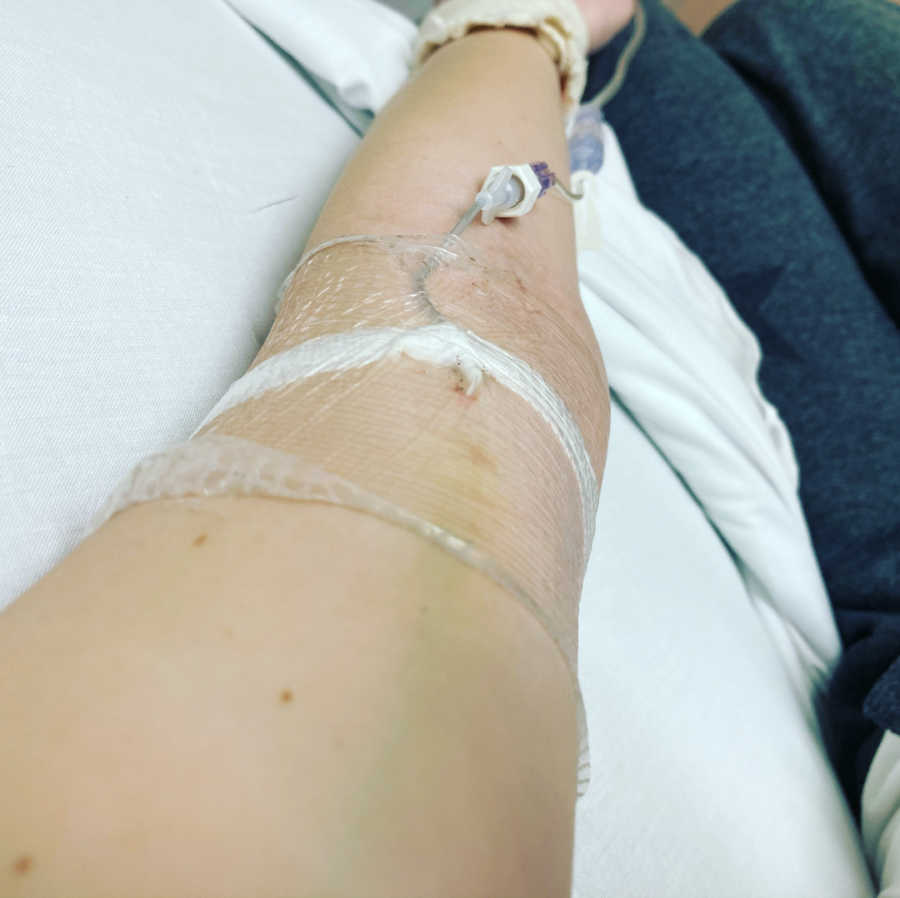
At first, my IVs were only a couple of times a year, but as time has gone on, they have increased in number. The last 2 years have been particularly brutal with courses often feeling like they almost run into each other. When I do IV antibiotics, I mostly do them myself at home. This requires me to mix up three IV antibiotics three times a day and push them directly into my veins through a long line. A long line looks like a cannula you may see in the back of a person’s hand in hospital. Unlike a cannula, it is soft and flexible and extends 8-22 centimeters under the skin, running usually from my wrist/elbow all the way up my arm.
When I first started doing home IVs, I found them to be hard work and tiring. Now, I have three small children to look after at the same time they are more exhausting than words can describe. IVs make me nauseous, exhausted, and upset my stomach to an almost apocalyptic level. And that’s when I don’t have an adverse reaction to them, something that is becoming a more and more frequent occurrence.
All though this may all sound extremely grim, my life, although sometimes a little challenging, is a very happy one. My husband and I are fortunate enough to have had three children, something neither of us ever thought would be possible. My first child, Ben, was born in 2015. When I initially told my consultant I wanted to have a baby, they were quick to warn me having a baby would pose a great risk to my health and getting pregnant may prove very difficult. Despite this, my whole CF team were very supportive of me when I decided to accept the risks and cared for me well during and after the pregnancy. Ben was a very easy baby; relaxed, happy and a good sleeper. John and I settled into parenthood naturally and all was well. However, when Ben was 9 months old, I accidentally fell pregnant again. Myself, John, and my CF team were all a little nervous at how I would cope with two so young, but again they looked after and supported me well.


At my 12-week scan, everything changed dramatically. The ultrasound showed I was carrying not one baby but two, and not only that but the twins were MCDA twins, meaning they shared a placenta. This is a rare type of identical twin. Sharing a placenta put the babies under a huge risk – they could start to mismanage the blood and nutrients coming through the placenta to them. If this happened, one would steal more from the other, potentially causing one to starve and the other to over-saturate themselves. This is called Twin to Twin Transfusion Syndrome and is sadly common and regularly fatal in MCDA twins. My already high-risk pregnancy became unimaginably more complicated. I began to see my obstetrician every other week for a scan and my CF team in the alternate week.
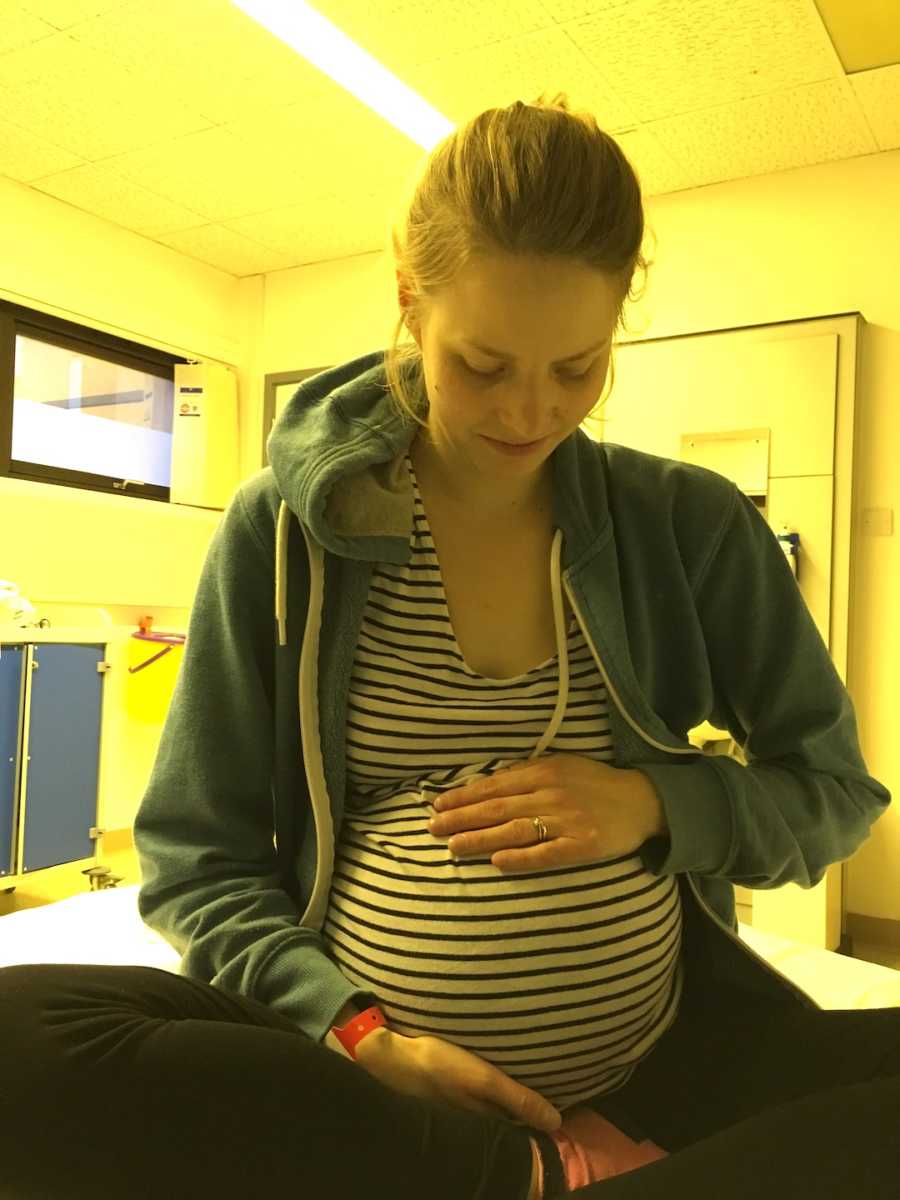
Despite this scary and stressful warning, the pregnancy went as smoothly as any twin pregnancy ever can, and before I knew where we were it was delivery day. I was induced, but the babies were so ready to come my labor (which had been under 3 hours for Ben) took a mere 23 minutes. The shock of such intense contractions caused my double-sized placenta to rip almost entirely from the wall of my womb, resulting in an almost fatal blood loss. Although their entry into the world was harrowing, the girls were able to come home only 10 days after being born, both still only 4 pounds. When I look back now at the photos of my three children all aged under 18 months, I really can’t believe we survived. At the time, we just had to get our heads down and push on, but now it seems like madness.


John is a self-employed plumber, having set up his own small business just days before we found out I was having twins. This does sometimes work to our advantage as he can leave the house a little later or return a little sooner on days when I really need him, but mostly it has led to us both be constantly over exhausted, with the stress of children, paperwork and responsibility weighing heavily on us. That being said, although these last 5 years have been incredibly taxing on both our relationship and my health, they have also been full of many wonderful moments. Neither of us could have imagined we would have three children so close together, or envision the joy that set up would bring. When we watch the children play together, we realize just how blessed we are, even when times are hard.


As I am getting older with my CF, life will become increasingly more difficult. Last year I turned 30, which in CF is something of a milestone. Although I have a good long number of years left and I certainly will try my best to stay as healthy as I possibly can to lengthen my days, the fact CF is a life limiting condition cannot be avoided. On average, patients reach 29-35 years of life, although hopefully with new treatments that average will steadily increase. This is an unpleasant truth, and one I came to terms with as a teenager. Having a heightened awareness of my own mortality could become depressing, but to me it has been a positive. I see my life as one that may well be short but will definitely be full of love and happiness. So far all is going well, and I hope will continue to do so for a long time to come.”

This story was submitted to Love What Matters by Abi Halstead from England, UK. You can follow her journey on Instagram. Submit your own story here, and be sure to subscribe to our free email newsletter for our best stories, and YouTube for our best videos.
Read more touching stories like this:
Provide beauty and strength for others. SHARE this story on Facebook with friends and family.